OpenMS is a flexible codebase that can be tailored to many different applications ranging from the standard label free analysis to top down, metabolomics, crosslinking or DIA.
Choose an application of OpenMS that you are interested in. The pages will provide explanations on how OpenMS can be used to solve your problems and link to workflows that allow you to apply the tools to your data.
If you cannot find your application in the menu on the left, more OpenMS tools can be found in the TOPP documentation.
Featured Tools
Intelligent data acquisition for top-down proteomics (FLASHIda)
FLASHIda is an intelligent online data acquisition algorithm for top-down proteomics (TDP) that ensures the real-time selection of high-quality precursors of diverse proteoforms. FLASHIda combines fast decharging algorithms in FLASHDeconv and machine learning-based quality assessment to identify optimal precursors for fragmentation. Currently the c# source code and instruction of FLASHIda is available in here under a BSD three-clause license. We are working on merging FLASHIda into OpenMS.
Quantification for Top-down Proteomics (FLASHDECONVQ)
FLASHDeconvQ performs MS1-level label-free quantification data analysis in top-down proteomics with an automatic coeluting proteoform resolution method.
To read more about it, please click here.
SRM Quantitation & QC (SMARTPEAK 2)
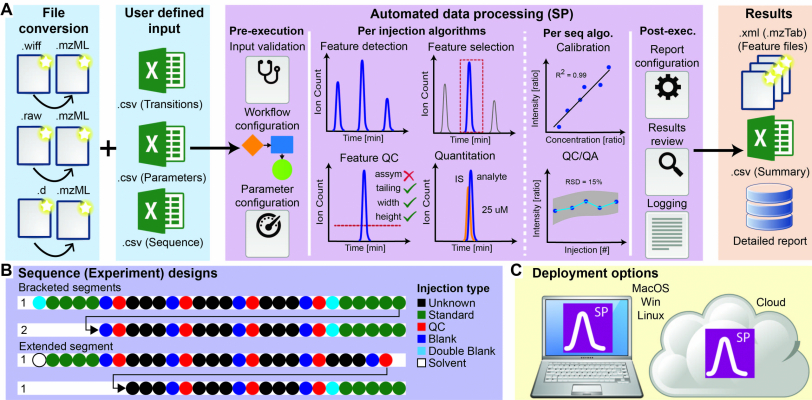
Based on OpenMS, a full SRM quantitation and quality control suite was developed by DTU BioSustain. It provides a standalone GUI for easy, fast, and scalable analysis of high-throughput metabolomics SRM experiments. It also added a set of state-of-the-art peak integration and peak alignment algorithms to the library.
Protein-nucleic acid cross-linking (OPENNUXL)
Thanks for your interest in OpenNuXL – our novel Protein-nucleic acid cross-linking search engine. It will be soon fully integrated into an official OpenMS release and all source code will be made available. Prior to publication, installers (and source code) are available on request: please contact sachsenb (at) informatik.uni-tuebingen.de
Efficient bayesian protein inference (EPIFANY)
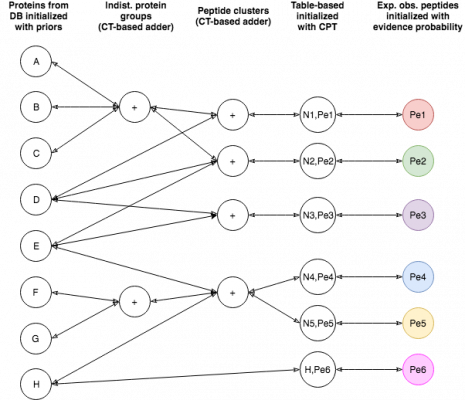
Introduction
EPIFANY is a protein inference engine based on a Bayesian network. Currently, a similar model to Fido is used with the main parameters alpha (pep_emission), beta (pep_spurious_emission) and gamma (prot_prior). If not specified, these parameters are trained based on their classification performance and calibration via a grid search by simply running with several possible combinations and evaluating. Unless you see very extreme output probabilities (e.g. many close to 1.0 or 0.0) or you know good parameters (e.g. from an earlier run), grid search is recommended, although slower. The tool will merge multiple idXML files (union of proteins and concatenation of PSMs) when given more than one. This is useful for fractions or replicates if they were not merged already. It assumes one search engine run per input file but might work on more with undefined behavior.
To read more about it, please click here.
Ultra fast MS1/MS2 deconvolution for top-down proteomics: FLASHDECONV 2.0 BETA+, finally with a GUI!
FLASHDeconv 2.0 beta+ with a GUI!!
Finally a GUI is here. You can find the GUI command in [OpenMS path]/bin folder. Go to [OpenMS path]/bin and run FLASHDeconvWizard! FLASHDeconv 2.0 beta+ works for MS1 and MS2 spectral deconvolution and feature deconvolution. It supports various output formats (e.g., *.tsv, *.mzML, *.msalign, and *.feature). FLASHDeconv 2.0 stable version will be officially integrated in OpenMS 2.7.0 released in near future. FLASHDeconv 2.0 beta+ also supports TopPIC identification better than the previous version, by generating all msalign and feature files for TopPIC inputs. We also added spectral merging function to support QTOF dataset analysis and NativeMS dataset analysis.
To read more about it, please click here.
Nucleic acid search engine (NASE)
NASE is now included in OpenMS release, 2.5.
To read more about it, please click here.
Protein-protein cross-linking (OPENPEPXL)
OpenPepXL: an open source peptide cross-link identification tool
OpenPepXL is a protein-protein cross-link identification tool implemented in C++ as part of OpenMS. It works with all uncleavable labeled and label-free cross-linkers but not (yet) with cleavable ones.
To read more about it, please click here.
RNA-Protien Cross-Linking (RNPXL)
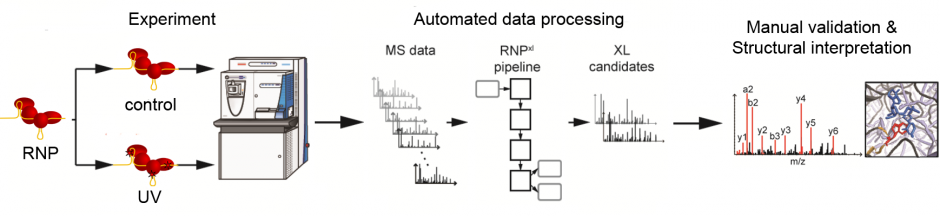
RNPxl has been integrated into OpenMS and also the Proteome Discoverer Community Nodes. No custom installer is required anymore.
To read more about it, please click here.
Stable isotope probing (METAPROSIP)
MetaProSIP: automated inference of elemental fluxes in microbial communities
To read more about it, please click here.
Featured workflows
Quantitative data independent proteomics (DIAPROTEOMICS)
DIAproteomics is a bioinformatics analysis pipeline used for quantitative processing of data independent (DIA) proteomics data.
To read more about it, please click here.
Data-independent acquisition metabolomics analyzer (DIAMETALYZER)
DIAMetAlyzer is a KNIME workflow which integrates DDA and targeted DIA analysis for metabolomics, which allows for false-discovery rate estimation based a target-decoy approach (see figure 1). It performs DDA based candidate identification and constructs a target/decoy assay library, which is used for DIA target extraction and statistical validation (FDR estimation).
To read more about it, please click here.
Quantification and identification of mhc peptides (MHCQUANT)
MHCquant: Identify and quantify peptides from mass spectrometry raw data
MHCquant is an analysis pipeline used for quantitative processing of data dependent (DDA) peptidomics data.
It was specifically designed to analyze immunopeptidomics data, which deals with the analysis of affinity-purified, unspecifically cleaved peptides that have recently been discussed intensively in the context of cancer vaccines. (https://www.nature.com/articles/ncomms13404)
To read more about it, please click here.